En los últimos años, un gran esfuerzo se ha dedicado al desarrollo de diversas técnicas moleculares para el diagnóstico de laboratorio de las micosis. En este contexto, la elucidación de la secuencia genómica completa de los principales patógenos fúngicos representa una oportunidad sin precedentes para los micólogos clínicos.
Además, existe una gran expectación sobre la posible aplicación de los rápidos avances en el campo de la genómica y la proteómica al estudio de la base molecular de la patogenicidad fúngica, la búsqueda de nuevos antifúngicos más eficaces y la identificación de antígenos fúngicos.
Introducción a la Investigación Micológica en la Era de la Genómica
Las técnicas tradicionales de diagnóstico micológico a menudo presentan problemas de falta de especificidad, sensibilidad, dificultades técnicas o de interpretación, y lentitud. Para solventar estos problemas, el desarrollo de técnicas inmunológicas y moleculares ha sido crucial.
Nos encontramos en el contexto de las llamadas "-ómicas", disciplinas científicas que abordan su objeto de estudio de manera global, como la genómica, proteómica, metabolómica y otros campos afines. El desarrollo a gran escala de las técnicas de la genómica y posgenómica ofrece una oportunidad sin precedentes para investigar los caracteres asociados a la patogenicidad de las especies fúngicas. También existe una gran expectativa sobre la posibilidad de aplicar estos avances al diagnóstico de laboratorio de las micosis y al diseño de nuevas estrategias terapéuticas más eficaces.
Técnicas de Diagnóstico Molecular de Hongos Patógenos
Hoy día, el micólogo clínico dispone de una amplia variedad de técnicas de diagnóstico molecular, cada una útil para determinar la identidad del agente etiológico en diferentes niveles taxonómicos. Es fundamental elegir la técnica adecuada en función del uso deseado: diagnóstico, epidemiológico o taxonómico.
Las técnicas moleculares para la detección e identificación de hongos patógenos se dividen en dos tipos:
- Métodos de amplificación de señal: Utilizan técnicas de hibridación de ácidos nucleicos. Las sondas de hibridación pueden confirmar la identificación de un cultivo o identificar el hongo patógeno en secciones tisulares, siendo habitualmente empleadas para patógenos como Histoplasma capsulatum y Coccidioides immitis.
- Métodos de amplificación de ácidos nucleicos: Se refieren fundamentalmente a todas las técnicas de reacción en cadena de la polimerasa (PCR). Existen técnicas de PCR desarrolladas para el diagnóstico de un gran número de micosis, como la aspergilosis y las dermatofitosis animales.
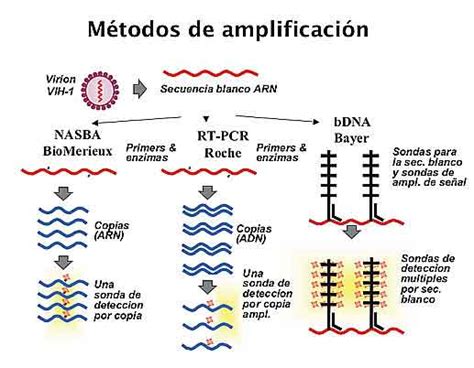
Además, existe un amplio abanico de técnicas para la tipificación molecular de aislamientos fúngicos, incluyendo las basadas en endonucleasas de restricción, amplificación de secuencias al azar, detección de secuencias cortas repetidas en tándem (minisatélites y microsatélites), y análisis de polimorfismos de base única. Estas técnicas suelen aplicarse al estudio epidemiológico de una infección y a la búsqueda de la fuente de contaminación por un patógeno, siendo también útiles para investigar la relación entre aislamientos de distintos pacientes y establecer la identidad de múltiples aislamientos de un mismo paciente.
La Investigación Micológica en una Nueva Era
Tanto el diagnóstico como la investigación micológica se dirigen hacia una nueva era, por al menos tres razones:
- Las infecciones fúngicas están cobrando importancia como causa de mortalidad en los países desarrollados. Por ejemplo, Candida spp. es el cuarto microorganismo más frecuentemente aislado en sangre de pacientes estadounidenses.
- La industria de los antifúngicos tiene una gran importancia económica para las empresas farmacéuticas, y la aparición de resistencias exige el conocimiento de su base genética.
- Se dispone de la secuencia de los genomas de los principales patógenos fúngicos (Aspergillus fumigatus, Candida albicans, Cryptococcus neoformans) y se trabaja en la secuenciación de muchos otros (Candida tropicalis, Pneumocystis jiroveci, Histoplasma capsulatum).

Otros factores que han contribuido a esta nueva concepción incluyen la mejora en las tecnologías de secuenciación y análisis de datos, que reducen costes y esfuerzo humano, y la disponibilidad de patrocinadores económicos para la investigación genómica en hongos. Por ello, la genómica, el estudio de toda la información genética de un organismo, tendrá un papel preponderante en los próximos años.
Hitos en la Genómica de los Hongos
La historia de la genómica en hongos es corta pero intensa. Esta carrera por descifrar los secretos del genoma fúngico comenzó en 1992 con la publicación de la secuencia completa del cromosoma III de Saccharomyces cerevisiae. Cuatro años después, en 1996, se hizo pública la secuencia completa de todo el genoma de esta especie de levadura. Estos eventos son considerados de gran relevancia, ya que representan el primer cromosoma y el primer genoma eucariota en secuenciarse.
Apareamiento en la levadura Saccharomyces cerevisiae, segunda parte
Posteriormente, se discutió la necesidad de estudiar en profundidad los genomas de hongos filamentosos. De estas discusiones surgieron proyectos piloto, como la secuenciación del cromosoma IV de Aspergillus nidulans. Sin embargo, no fue hasta el año 2000 cuando la genómica de los hongos filamentosos comenzó a despegar con el proyecto Fungal Genome Initiative (FGI). En esta iniciativa participaron diversos miembros de la comunidad científica y consorcios de secuenciación, destacando el Whitehead Institute for Biomedical Research (ahora Broad Institute). Finalmente, en 2003 se hizo pública la secuencia completa del genoma de Neurospora crassa, el primero correspondiente a un hongo filamentoso.
La Iniciativa del Genoma Fúngico (FGI)
El proyecto Fungal Genome Initiative (FGI) ha sido un gran impulso para la genómica de los hongos filamentosos. Su filosofía y líneas principales están recogidas en varios documentos. Desde su inicio en el año 2000, diversas instituciones y consorcios de secuenciación se han sumado, destacando la participación del Broad Institute como "centro neurálgico", colaborando activamente en la secuenciación de genomas de gran interés como A. nidulans, A. terreus, C. albicans, C. immitis y C. neoformans. La coordinación entre todas las instituciones implicadas es indispensable en un proyecto de tal magnitud.
La propuesta inicial del FGI incluyó cinco especies de hongos con importancia para la salud humana (C. neoformans serotipo A, Coccidioides posadasii, P. jiroveci, Trichophyton rubrum y Rhizopus oryzae), además de especies con importancia económica o como organismos modelo. Las especies fúngicas se eligieron siguiendo criterios como su impacto en la salud o economía humana, su valor como herramienta para estudios de genómica comparada, la existencia de recursos genéticos y el interés de la comunidad científica.
La gran aportación de esta iniciativa ha sido abordar los proyectos genoma de hongos desde una perspectiva taxonómica. Se considera que una selección equilibrada de los organismos fúngicos a secuenciar maximizaría el valor de los datos obtenidos, permitiendo estudios evolutivos y de genómica comparada, lo que contrasta con los pequeños proyectos independientes anteriores a esta iniciativa.
Situación Actual de los Proyectos Genoma en Hongos
Actualmente, se conoce la secuencia de más de 18 especies de hongos. En los primeros años de la genómica en hongos, las levaduras centraron la atención, pero ahora la mayoría de los proyectos de secuenciación en curso se centran en los hongos filamentosos.
También están cobrando importancia proyectos que no buscan una secuenciación completa, sino la identificación de los genes que se expresan en un organismo en un momento determinado, conocidos como etiquetas de secuencia expresada (EST). Esta aproximación permite identificar las regiones codificantes del genoma que se expresan bajo determinadas condiciones, como los genes que expresa un hongo patógeno dentro de un hospedador, entre los cuales pueden encontrarse posibles factores de virulencia.
Genómica Funcional
La secuenciación genómica de microorganismos se ha vuelto casi sistemática y no representa un desafío en sí misma. La mayor complejidad radica en interpretar la información obtenida. La secuencia de un genoma es similar a un diccionario sin definiciones; se trata de buscar marcos abiertos de lectura (ORF), identificar la función de cada gen y sus posibles interacciones. En general, es más difícil identificar ORF en hongos filamentosos que en levaduras, debido a la mayor complejidad de la estructura de sus genes y al menor conocimiento sobre los mecanismos de regulación de su expresión.
Existen dos formas básicas de abordar la genómica funcional:
- Análisis de patrones de expresión génica: Se recurre a los chips de ADN.
- Análisis de la función de los genes: Se anula la función por mutación.
Arrays de ADN en el Estudio de la Virulencia Fúngica
Un array es un conjunto de sondas moleculares fijadas de manera ordenada sobre un soporte sólido. Cuando estas sondas son moléculas de ADN, se habla de arrays de ADN. Esta técnica se basa en la hibridación específica entre un ARN mensajero (ARNm) y la molécula de ADN de la cual deriva por transcripción.
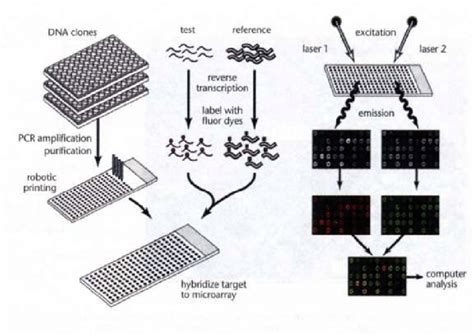
La tecnología de arrays o chips de ADN se utiliza cada vez con más frecuencia para estudiar simultáneamente y de manera cuantitativa la expresión de genes a escala genómica. En estudios de patogenicidad, la idea subyacente es que la identificación de los patrones de expresión génica del hongo patógeno bajo condiciones ambientales y condiciones asociadas a una infección puede ayudar en la búsqueda de potenciales factores de virulencia.
Por ejemplo, el estudio de la expresión génica en Aspergillus fumigatus durante los procesos infecciosos mediante chips de ADN permitirá ampliar los conocimientos sobre la virulencia de este hongo. Un estudio examinó los perfiles de expresión génica en A. fumigatus a 30, 37 y 48 °C. Se observó que a 37 °C, se expresa un mayor número de genes relacionados con la patogenicidad (p. ej., genes que codifican proteínas que responden al estrés oxidativo o al sistema inmunitario del hospedador y genes implicados en la producción de toxinas) que a las otras dos temperaturas, incluso sin contacto con células de mamíferos.
Análisis de Mutantes en la Genómica Funcional
El análisis de mutantes de deleción para inferir la función de los genes y realizar cartografías cromosómicas es una técnica tradicional en genética. En la era de la genómica, se busca obtener y analizar mutantes de forma masiva. Tras generar colecciones de mutantes, por ejemplo, por transposición, se realiza un cribado para encontrar microorganismos incapaces de realizar alguna función particular.

Genómica Comparada de Levaduras y Virulencia
El grupo de investigación de Genómica Funcional de Levaduras (GFL) de la Universitat de València ha descubierto que cuando una célula se divide en dos de diferente tamaño (división asimétrica), se genera un problema en el reparto de algunos componentes celulares que no se da en las células que se dividen simétricamente. En el caso del reparto de genomas, es inevitable dar a cada célula hija la misma cantidad. Utilizando como modelo de trabajo la levadura de la cerveza (Saccharomyces cerevisiae), que tiene una división asimétrica por gemación, los investigadores han comprobado que este problema podría llevar a un desajuste catastrófico en algunos procesos moleculares incompatible con la supervivencia de la especie. La evolución ha modelado de forma diferente la regulación global de la síntesis de RNA en las células con división simétrica o asimétrica para ajustar cada una a sus necesidades.
El grupo GFL lleva años trabajando en metodologías para determinar cuantitativamente las tasas de transcripción y degradación de las moléculas de ARN mensajero (mRNA) a escala genómica, centrándose en el estudio de los mecanismos de la regulación génica, utilizando Saccharomyces cerevisiae como modelo experimental.
Origen de la Duplicación del Genoma en Levaduras
Investigadores del Centro de Regulación Genómica (CRG) proponen una nueva teoría que explicaría el origen de la duplicación del genoma al inicio del linaje de las levaduras. Las levaduras son hongos unicelulares que se originaron hace más de 100 millones de años, conocidos por su capacidad de fermentar hidratos de carbono y su uso en bebidas y alimentos, además de ser un organismo modelo en investigación.
La levadura Saccharomyces cerevisiae fue el primer organismo eucariota en ser secuenciado en los años 90, y desde entonces se ha aceptado que su genoma había sufrido una duplicación entera, un fenómeno que también se ha observado en el origen de los vertebrados, en humanos y en especies vegetales.
La científica Marina Marcet-Houben y el jefe de grupo del CRG Toni Gabaldón han estudiado el origen de la duplicación del genoma entero en las levaduras. Su trabajo, publicado en PLOS Biology, demuestra que la aparición de genes duplicados no fue causada por una simple duplicación del genoma entero, sino que fue fruto de la hibridación entre dos especies diferentes. Esta propuesta contradice la teoría más establecida y ofrece nuevos puntos de vista sobre un proceso clave para comprender la evolución de los genomas y de las especies.
Utilizando metodología computacional de última generación y combinando datos genómicos con una herramienta diseñada por el grupo del Dr. Gabaldón, capaz de reconstruir duplicaciones de genes y datar su aparición evolutiva, se encontró que la edad de los genes duplicados era mucho mayor de lo que predecía la teoría de la duplicación simple del genoma. Los datos indicaban que los genes duplicados habían comenzado a divergir mucho antes, lo que abrió la posibilidad de una hibridación entre especies.
La hipótesis de la hibridación implica que no son necesarios cambios masivos y rápidos para originar nuevas funciones a partir de secuencias duplicadas, ya que la hibridación combina propiedades de los dos linajes parentales desde el primer día, abriendo nuevas oportunidades ecológicas y evolutivas. Esta propuesta se refuerza al observar que el número de duplicaciones de genoma es mucho mayor en plantas cultivadas, donde la hibridación forzada ha sido clave para mejorar la producción.
Saccharomyces cerevisiae como Patógeno Oportunista Emergente
Aunque Saccharomyces cerevisiae es ampliamente utilizada biotecnológicamente, como suplemento dietético y probiótico, y se considera "Generalmente Reconocida como Segura" (GRAS), actualmente se le clasifica como un patógeno oportunista emergente de baja virulencia, capaz de causar infecciones principalmente en hospedadores inmunodeprimidos.
Para conocer el posible papel de esta levadura como patógeno emergente, se obtuvieron cepas de S. cerevisiae de 8 productos dietéticos comerciales. Se aplicaron técnicas moleculares de identificación (análisis de restricción de la región ribosomal 5.8S-ITS) y caracterización (análisis de restricción del DNA mitocondrial y amplificación por PCR de elementos delta). Se identificaron 6 aislados como S. cerevisiae y 2 como híbridos interespecíficos (S. cerevisiae x S. kudriavzevii y S. cerevisiae x S. bayanus). La caracterización reveló que 4 de estos aislados eran la misma cepa, resultando en 5 aislados de levadura diferentes.
Estudio de Rasgos Fenotípicos y Virulencia in vivo
Se llevó a cabo un estudio de rasgos fenotípicos in vitro asociados con la virulencia en levaduras patógenas, como el crecimiento a altas temperaturas, secreción de enzimas extracelulares (proteasas y fosfolipasas), crecimiento pseudofilamentoso y crecimiento invasivo. Los aislados híbridos mostraron negatividad para la mayoría de estos rasgos, mientras que los aislados D2, D4 y D14 presentaron estos rasgos con un nivel alto a moderado, indicando una posible capacidad infectiva.
Posteriormente, se realizó un estudio de infección sistémica in vivo en ratones inmunocompetentes BALB/c, estudiando el alojamiento de estas levaduras en cerebro y riñón a los 7, 15 y 30 días postinfección. El aislado híbrido D23 mostró incapacidad para colonizar ambos órganos, mientras que los aislados D2, D4 y D14 mostraron alojamiento en cerebro y riñón. El aislado D14 fue el que mostró mayor potencial patógeno, causando mortalidad en ratones.

Se estableció un modelo murino de infección gastrointestinal para valorar la capacidad de translocación intestinal y diseminación de cepas de S. cerevisiae, utilizando el aislado virulento D14 y el avirulento D23. Ambos aislados fueron capaces de atravesar la barrera intestinal, pero solo el aislado virulento D14 mostró diseminación consistente en órganos distantes como hígado, riñones, cerebro y nódulos mesentéricos.
Análisis Genómico y Transcriptómico de Cepas de S. cerevisiae
Se realizó un análisis genómico de 4 cepas de S. cerevisiae: el aislado D14, una cepa de laboratorio avirulenta W303 y dos aislados clínicos virulentos (60 y 102). Se observó variabilidad en la composición genómica de las zonas subteloméricas, siendo mayor en los aislados virulentos. La mayoría de los genes con variación en el número de copias se situaban en estas regiones. También se encontró aneuploidía en el aislado clínico 60, lo que podría proporcionar una ventaja selectiva en ciertas condiciones ambientales. Se detectaron cambios en el número de copias de genes relacionados con la reparación de bases causada por daño oxidativo o genes del metabolismo del glutatión.
Un estudio comparativo de la supervivencia en sangre humana de las cepas virulentas D14 y 60 y la cepa de laboratorio avirulenta W303 demostró que las cepas virulentas fueron capaces de sobrevivir en sangre humana, con un porcentaje de supervivencia equivalente al de una cepa patógena de C. albicans.
Utilizando el modelo de supervivencia en sangre humana, se llevó a cabo un análisis transcriptómico de los aislados virulentos D14 y 60, la cepa de laboratorio W303 y una cepa vínica avirulenta CECT 10.431. Se observó una respuesta transcripcional específica por parte de los aislados virulentos (D14 y 60) de genes implicados en la biosíntesis y metabolismo de aminoácidos y en genes de respuesta al estrés oxidativo. Además, tras la aplicación de un estrés oxidativo, las cepas virulentas mostraron mayor resistencia que las cepas avirulentas (CECT 10.431 y W303), sugiriendo una correlación entre la resistencia al estrés oxidativo y la patogenicidad. Los cambios observados en el análisis genómico no pudieron correlacionarse con el nivel de expresión de estos genes en el análisis transcriptómico. Por lo tanto, una expresión diferencial entre cepas virulentas y avirulentas podría ser resultado de la adaptación de las primeras al medio sanguíneo. Este estudio es de gran interés para las industrias alimentarias que incluyen S. cerevisiae en sus preparados, con el fin de evitar la administración de dichas cepas de levaduras a individuos inmunodeprimidos.
Ingeniería Genética en Levaduras: Cromosomas Sintéticos
La levadura natural es un organismo importante para la fabricación de medicamentos, cerveza y biocombustibles, y su función podría mejorar con el diseño de cromosomas sintéticos y sustituibles para producir mejores versiones de las materias primas, lo que implicaría crear antibióticos nuevos o combustibles más eficientes con el medioambiente.
Un consorcio internacional de más de 200 científicos, el Proyecto Levadura Sintética, logró crear un cromosoma artificial de levadura en marzo de 2014. Ahora, se han sumado cinco cromosomas sintéticos más, lo que representa más de un tercio del genoma total de la levadura. Estos resultados son un gran avance hacia la creación del primer organismo completamente sintético, con la expectativa de completar los 16 cromosomas de la levadura a finales de este año.
Las células de la levadura con su genoma completo sintético, denominado Saccharomyces cerevisiae 2.0 o Sc2.0, serán muy valiosas para aplicaciones académicas e industriales. El diseño del genoma de Sc2.0 será un 8% más pequeño que el genoma de la levadura natural, eliminando ADN "basura" no codificante y otras secuencias genéticas reubicadas que pueden hacer que el ADN sea inestable y propenso a mutaciones. A pesar de estos cambios, las células de levadura con los cromosomas alterados crecieron con normalidad, sugiriendo que los investigadores podrán realizar cambios de mayor alcance, explorando los límites de la ingeniería genética para conseguir que la levadura genere productos aún más útiles.
Estos resultados ayudan a los científicos a comprender mejor los componentes genéticos necesarios para la vida y abren el camino a una nueva era de terapia génica, donde se podrían proporcionar no solo un único gen, sino redes enteras de genes humanos para fines terapéuticos.
Levaduras en la Fermentación Alcohólica: Identificación y Tipado Molecular
La fermentación alcohólica del vino es un proceso biológico intencionado en el que las levaduras metabolizan azúcares del mosto de uva, transformándolos en etanol, dióxido de carbono y otros productos secundarios. La comunidad microbiana del mosto es muy diversa y depende de factores como el tipo de uva, las condiciones climáticas, las características del suelo y las prácticas agrícolas.
Inicialmente, predominan levaduras no Saccharomyces, clasificadas en estrictamente aerobias (Pichia, Rhodotorula, Cryptococcus), con capacidad fermentativa limitada (levaduras apiculadas como Kloeckera) y con metabolismo fermentativo típico pero baja tolerancia al etanol (Metschnikowia, Kluyveromyces, Zygosaccharomyces, Torulaspora). A medida que aumenta la concentración de etanol (alrededor del 3-4% v/v), estas especies son gradualmente sustituidas por levaduras del género Saccharomyces, siendo S. cerevisiae la principal. Es una simplificación, ya que la ecología microbiana en la fermentación alcohólica es compleja, identificándose hasta 40 especies diferentes de S. cerevisiae en el mosto de uva.
La elección adecuada de cepas de levaduras es clave para optimizar la seguridad y calidad en la elaboración de vinos. Esta selección se basa en fermentaciones espontáneas, aislando cepas autóctonas con elevada capacidad fermentativa y resistencia al etanol y al estrés. La identificación precisa de las especies presentes en el mosto y durante la fermentación permite mejorar el control microbiológico, seleccionar cepas starter y evitar contaminaciones. Tradicionalmente, la identificación y diferenciación de levaduras se realizaba mediante criterios morfológicos y fisiológicos.
Genoma de Levaduras y Marcadores Moleculares
El material genético de las levaduras se distribuye en tres componentes principales: el genoma nuclear, el mitocondrial y los plásmidos presentes en el citoplasma. En Saccharomyces cerevisiae, el DNA nuclear representa aproximadamente el 80% del contenido genético total en su estado diploide, compuesto por unos 20.000 kilobases (kb), organizados en 32 cromosomas lineales (16 pares homólogos). El cromosoma XII es el más extenso y variable entre cepas, albergando las secuencias del DNA ribosómico (rDNA), con genes responsables de la síntesis de las subunidades del RNA ribosómico (25S, 18S, 5.8S y 5S).
El tipado molecular permite discriminar especies y cepas de levaduras a partir de secuencias de DNA, patrones de restricción, perfiles proteicos o huellas genéticas. Estas herramientas son esenciales para estudios de biodiversidad, ecología microbiana, trazabilidad y selección industrial. Las técnicas moleculares ofrecen la ventaja de analizar directamente el genoma, sin verse afectadas por el estado fisiológico de las células, proporcionando resultados más fiables y aplicables.
En el análisis molecular de levaduras, dos regiones ribosomales se emplean comúnmente para su identificación:
- El dominio D1/D2 de la subunidad grande del ribosoma (LSU), útil para especies ascomicetas.
- La región que abarca los espaciadores transcritos internos junto con el gen ribosomal 5.8S (ITS1-5.8S-ITS2), que permite identificar tanto ascomicetas como basidiomicetas.
Los genes ribosómicos 18S, 5.8S y 26S se disponen en tándem formando unidades de transcripción, dentro de las cuales se encuentran dos regiones conocidas como ITS1 e ITS2, que flanquean el gen 5.8S. Estas regiones, denominadas “espaciadores internos transcritos” (ITS), aunque se transcriben, no forman parte del RNA funcional. Su alta variabilidad genética ha hecho que sean ampliamente utilizadas como marcadores moleculares para diferenciar géneros y especies de levaduras.
Técnicas Moleculares para la Identificación y Caracterización de Levaduras
PCR-RFLP de ITS
La técnica comúnmente utilizada para analizar las regiones ITS es la PCR de los ITS y su posterior RFLP (polimorfismo de longitud de fragmentos de restricción tras amplificación por PCR). El procedimiento comienza con la amplificación de la región ITS mediante los cebadores ITS1 e ITS4. Posteriormente, el producto amplificado se digiere con enzimas de restricción, como CfoI, HaeIII, HinfI, DdeI o MboI. Estos patrones pueden compararse con bases de datos como GenBank para la identificación precisa de las levaduras. Una de las principales ventajas de este método es que permite trabajar directamente con cultivos celulares, sin necesidad de purificar previamente el DNA.
Secuenciación Sanger de LSU
Entre las metodologías basadas en PCR disponibles, la secuenciación por tecnología Sanger de la región LSU ha demostrado ser la más confiable para la identificación a nivel de especie. Para amplificar el dominio D1/D2, se utilizan con frecuencia los cebadores NL-1 y NL-4. En ciertos clados, es posible relacionar agrupaciones evolutivas con aplicaciones biotecnológicas.
En algunos grupos taxonómicos, la secuenciación de una única región no es suficiente para discriminar entre especies cercanas, lo que ha impulsado el uso de enfoques multigénicos que combinan regiones ribosomales con genes codificantes, tanto del núcleo como de la mitocondria.
Análisis del DNA Mitocondrial (mtDNA)
El DNA mitocondrial de las levaduras está compuesto por una única molécula circular de doble cadena con una longitud aproximada de entre 75 y 85 kilobases (kb). En 1992, Querol y colaboradores desarrollaron un método que permitió analizar el DNA mitocondrial (mtDNA) sin necesidad de utilizar gradientes de cesio ni realizar la purificación previa de las mitocondrias. La base de esta técnica radica en la diferencia en la composición de bases nitrogenadas entre el DNA mitocondrial y el nuclear. El mtDNA presenta un alto contenido en adenina y timina (A+T), alcanzando aproximadamente un 75%, mientras que su proporción de guanina y citosina (G+C) es baja, cercana al 20%. Cuando se realiza una digestión enzimática sobre DNA total, las enzimas que reconocen secuencias como GANTC o GGCC producirán pocos cortes en el mtDNA, permitiendo que sus fragmentos se visualicen claramente como bandas definidas en geles de agarosa. Es importante señalar que los patrones obtenidos en la electroforesis dependen tanto de la enzima utilizada como de la especie de levadura analizada. En el caso de Saccharomyces cerevisiae, las enzimas de restricción más recomendadas son Hinf I y Hae III. En este tipo de análisis, Hinf I se emplea con frecuencia debido a que existe una base de datos amplia sobre los patrones generados con esta enzima, lo que facilita la interpretación y comparación de resultados.
Electroforesis en Campo Pulsado (PFGE)
El DNA genómico de las levaduras puede ser analizado mediante PFGE, una técnica que implica su digestión con enzimas de restricción de corte poco frecuente, seguida de su separación en un gel de agarosa usando electroforesis en campo pulsado. Este método aplica pulsos eléctricos alternos desde distintos ángulos, lo que permite redirigir continuamente la migración de las moléculas de DNA, evitando que queden atrapadas en la matriz del gel. Gracias a esta técnica, es posible resolver fragmentos de DNA superiores a los 225 kilobases (kb). Para garantizar la precisión en la interpretación de los geles, se utilizan estándares cromosómicos comerciales de Saccharomyces cerevisiae.
La PFGE permite detectar reordenamientos cromosómicos, como translocaciones, deleciones o amplificaciones, característicos del elevado grado de polimorfismo observado en el genoma de las levaduras. Estas variaciones estructurales entre cepas se manifiestan como diferencias de tamaño en los cromosomas. La cariotipificación mediante PFGE también se ha empleado para estudiar cepas implicadas en fermentaciones vínicas, así como para la identificación de Dekkera bruxellensis en vinos. En S. cerevisiae, los perfiles polimórficos generados por PFGE reflejan eventos evolutivos como inserciones o deleciones de fragmentos largos de DNA en cromosomas homólogos. Aunque esta técnica ofrece gran resolución en la caracterización de cepas individuales, su aplicabilidad es limitada frente a un amplio espectro de especies contaminantes, debido a los polimorfismos interespecíficos.
RAPD-PCR
La técnica RAPD-PCR permite amplificar aleatoriamente fragmentos del DNA genómico utilizando cebadores de secuencia arbitraria, generando patrones únicos de bandas tras su visualización por electroforesis, conocidos como “huellas digitales”. Esta herramienta es útil para detectar diferencias genéticas entre especies o cepas de levaduras, especialmente en fermentaciones espontáneas. Mediante esta metodología, se pueden seleccionar levaduras autóctonas con características deseables para la vinificación. La técnica RAPD-PCR también ha sido empleada para analizar la variabilidad genética dentro del grupo Saccharomyces sensu stricto, diferenciando entre especies muy relacionadas como S. cerevisiae, S. bayanus, S. paradoxus y S. pastorianus, así como entre grupos intraespecíficos. Gracias a su carácter multilocus, RAPD permite detectar el origen híbrido de algunas especies, identificando fragmentos de DNA compartidos entre híbridos y sus cepas parentales. Aunque su principal ventaja es que no requiere información previa sobre la secuencia del DNA y puede revelar polimorfismos a lo largo de todo el genoma, la baja temperatura de hibridación hace que sus resultados sean poco reproducibles, por lo que se necesitan múltiples repeticiones y el uso de varios iniciadores para mejorar la resolución.
AFLP (Amplified Fragment Length Polymorphism)
La técnica AFLP se presenta como una herramienta poderosa para el análisis genético y la caracterización de levaduras, especialmente útil para discriminar entre cepas a un nivel de resolución superior al de otras técnicas como los RAPD. Al igual que los RAPD, los AFLP no requieren información previa sobre la secuencia del genoma para el diseño de los cebadores, lo que representa una ventaja importante en el estudio de organismos con genomas poco caracterizados. El procedimiento de AFLP implica varias etapas: digestión del DNA genómico con enzimas de restricción, comúnmente una combinación de enzimas de corte frecuente y poco frecuente (por ejemplo, EcoRI y MseI), y posterior ligación de adaptadores específicos a los extremos de los fragmentos.
Apareamiento en la levadura Saccharomyces cerevisiae, segunda parte
tags: #genomica #comparativa #con #levadura #virulencia